
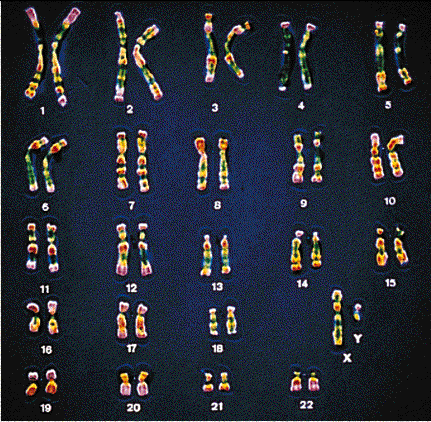
“Une maladie rare est une affection touchant un nombre restreint de personnes (faible prévalence), à savoir moins de une personne sur 2000 selon le seuil admis en Europe ; en France, on dit qu’une maladie est rare si moins de 30 000 personnes en sont atteintes. On dénombre environ 7000 maladies rares dont 80% sont d’origine génétique, mais chaque semaine, de nouvelles maladies rares sont définies. Les maladies rares concernent 3 à 4 millions de personnes en France, et près de 25 millions en Europe. Un grand nombre de ces pathologies sont aussi dites «orphelines» parce que les populations concernées ne bénéficient pas de réponse thérapeutique”
Le syndrome PhelanMcDermid ( SPM ) aussi appelé syndrome de la délétion 22q13 est un syndrome génétique rare causé par l’absence du gène SHANK3/ProSAP2 sur l’extrémité terminale du chromosome 22, une mutation du gène SHANK3 ou un chromosome 22 en anneau .
La forme la plus commune de PMS est causée par une délétion chromosomique de novo mais un des parents peut également être porteur.
A Lire => Un document essentiel réalisé par Orphanet en collaboration avec l’association fcse du syndrome Phelan-mcdermid :
https://www.orpha.net/data/patho/Han/Int/fr/PhelanMcDermid_FR_fr_HAN_ORPHA48652.pdf
Les causes : Le gène SHANK3 est situé dans le segment 22q13 du chromosome et est considéré comme le gène le plus important dans le déclenchement du SPM. Certaines personnes atteintes de syndrome n’ont pas de délétion, mais ont plutôt une “faute d’orthographe” dans le gène SHANK3, appelée mutation. Les mutations sont généralement de nouveaux événements et ne sont pas héritées d’un parent.
Des gènes comme SHANK3 enseignent au corps comment il doit se former et fonctionner. Lorsque le matériel génétique est altéré, qu’il s’agisse d’une délétion ou d’une mutation, il peut empêcher le gène de fonctionner correctement. Le corps reçoit alors un ensemble différent d’instructions, et fonctionne donc différemment. Ces différences constituent les symptômes de la maladie.
Environ 20% des personnes atteintes de SPM présentent une délétion, voire une duplication, causée par une translocation chromosomique. Une translocation se produit quand un ou plusieurs «bras» de différents chromosomes se détachent et changent de place. De plus, d’autres personnes atteintes de SPM ont ce qu’on appelle un chromosome en anneau, ou anneau 22, ainsi appelé parce que les extrémités très distales du chromosome 22 se détachent et les «nouvelles extrémités» du chromosome collent les unes aux autres pour créer un chromosome circulaire.
Les caractéristiques les plus communes chez les personnes atteintes du syndrome sont un retard de développement, une déficience intellectuelle à des degrés divers, un retard ou une absence de langage, des symptômes du trouble du spectre autistique, un faible tonus musculaire, des retards moteurs et des problèmes épileptiques. Les troubles du sommeil sont également fréquemment signalés, tout comme les troubles intestinaux chroniques ou les reflux gastro-oesophagien.
Moins fréquemment, certains enfants présentent des malformations cardiaques ou rénales. Ce sont souvent ces premiers symptômes perceptibles qui incitent les familles à amorcer le processus de diagnostic.
Cependant, il faut bien garder en tête que chaque personne est différente, même celles porteuses du syndrome !
Les symptômes ne seront donc pas les mêmes chez toutes les personnes et ce n’est pas parce qu’une personne n’arrive pas à faire telle ou telle chose qu’une autre ne sera pas capable de le faire. Par ailleurs, les apprentissages évoluent dans le temps et ne sont pas figés à un instant T. Chaque être humain est unique et si son évolution est liée en partie à son patrimoine génétique, elle ne dépend pas que de cela. Son caractère propre, l’ environnement socio-culturel et géographique dans lequel elle vit, les soins et l’éducation qu’elle reçoit, l’entourage et l’affection qu’on lui porte jouent également un rôle primordial.
Syndrome Phelan-mcdermid et autisme : Environ 75% des personnes porteuses du syndrome ont reçu un diagnostic de trouble du spectre de l’autisme. Par ailleurs, on estime que 1% des personnes autistes ont le syndrome de Phelan-McDermid. Cela signifie qu’entre 1/8 000-15 000 (y compris les délétions 22q13.3 et les variants du gène SHANK3) ont le syndrome. Cependant, cela peut être une sous-estimation puisque tous les patients atteints de SPM ne présentent pas d’autisme.
En France et dans le monde :
A l’heure actuelle, un peu plus de 1400 cas ont été diagnostiqués à travers le monde (janvier 2017) et près de 200 en France. Cependant, ceci ne concerne évidemment que les personnes ayant reçu un diagnostic donc le nombre de personnes porteuses est largement supérieur.
La plupart des cas identifiés sont les petits enfants parce que les tests se font généralement tôt dans la vie et un test fiable n’a pas commencé avant 1998. Par conséquent, la plupart des informations sur le syndrome Phelan-mcdermid concernent les enfants et il y a moins d’ expériences d’adultes atteints de SPM. Il est donc important que les parents d’un adulte qui soupçonnent un SPM effectuent un dépistage.
Faire le diagnostic : Le diagnostic de SPM doit être pris en compte chez les personnes ayant une déficience intellectuelle avec ou sans autisme et / ou présentant des caractéristiques physiques atypiques. Comme l’absence de développement de la parole est une caractéristique courante du syndrome, ce diagnostic doit également être envisagé chez tout enfant présentant un retard prononcé de la parole. Le SPM devrait également être pris en compte dans le diagnostic différentiel chez les personnes ayant des antécédents d’hyponatrémie néonatale de cause inconnue.
Un type de test génétique appelé biopuces (analyse de l’expression génétique à grande échelle) est la méthode la plus courante pour diagnostiquer le syndrome de Phelan-McDermid. Les principaux types de technologie de puces à ADN comprennent l’aGHG (hybridation génomique comparative), et l’analyse des polymorphismes nucléotidiques (SNP) . Ces deux tests trouveront une perte ou un gain de matériel chromosomique incluant de très petits changements. Cela signifie qu’ils peuvent détecter des pertes ou des gains résultant de délétions ou de duplications simples, d’une translocation déséquilibrée, de chromosomes en anneau et de tout changement structurel entraînant une perte / gain de matériel. Cependant, ces tests ne peuvent pas «voir» la structure du chromosome, ils ne peuvent donc pas dire si une translocation ou un chromosome en anneau est présent.
Hybridation in situ par fluorescence (FISH) ou l’analyse chromosomique (caryotype). FISH ou un caryotype est nécessaire pour identifier les translocations déséquilibrées ou le chromosome de l’anneau. FISH ou caryotype est également utilisé pour étudier les parents d’individus avec des translocations déséquilibrées pour déterminer si l’un des parents porte une forme équilibrée de la translocation qui est vu dans leur enfant. Des tests de séquençage ADN peuvent détecter des mutations dans le gène SHANK3.
Le registre international du syndrome de Phelan-McDermid (PMSIR) mis en place par la Fondation américaine du syndrome contient la plus grande base de données de cas diagnostiqués de syndrome Phelan-mcdermid dans le monde. Toute personne ayant un changement génétique dans la région 22q13 est encouragée à s’inscrire sur ce registre afin de se faire recenser. http://www.pmsf.org/membership/pmsf-membership_test-embed_french
Que faire après le diagnostic ?
Les personnes porteuses du syndrome sont prises en charge en fonction des symptômes qu’elles présentent, qui ne seront pas forcément les mêmes pour toutes.
Il n’existe actuellement aucun remède ou traitement spécifique au syndrome mais de nombreuses thérapies/prises en charge permettent d’aider efficacement les personnes concernées et de les faire progresser.
Votre médecin, pédiatre, neuro-pédiatre doit être votre interlocuteur privilégié pour mettre en place le suivi optimal.
En raison des problèmes parfois multiples, une équipe de plusieurs spécialistes au niveau médical et/ou éducatif peut être nécessaire pour aborder les différents domaines de préoccupation.
Au niveau médical, il est conseillé dans un premier temps de :
- Avoir une échographie rénale et un échocardiogramme dans les 3 mois suivant le diagnostic.
- Contrôler l’ouïe et la vision dans les 6 mois suivant le diagnostic, mais dans les 3 mois si l’enfant a moins de 4 ans ou si les parents ont des problèmes de vision ou d’ouïe.
- Faire des évaluations développementales et neurologiques dans les 6 à 12 mois.
- Un neurologue peut alors demander un EEG et une IRM, si nécessaire.
Les informations fournies ici sont destinées à améliorer et non à remplacer, la relation directe entre le patient et les professionnels de santé. Elles ne peuvent donc en aucun cas remplacer une consultation médicale.
Pour toute question sur la délétion 22q13, consulter votre médecin et/ou généticien.






